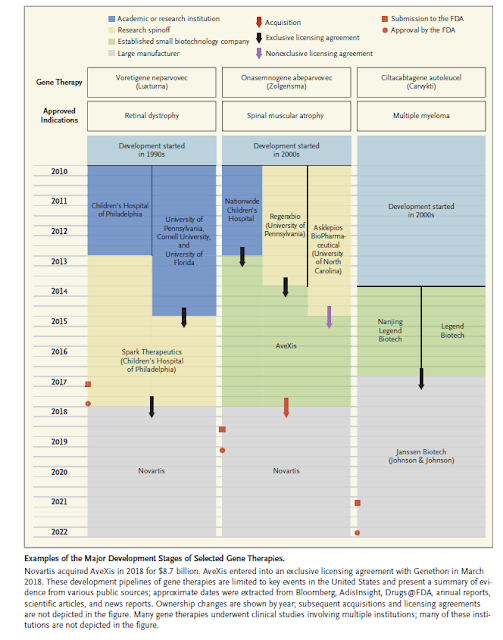
High drug prices are not justified by industry’s spending on research and development
Les dues retòriques per justificar els preus alts dels medicaments són: cal recuperar la inversió en recerca, i cal relacionar el preu amb el valor que aporten. La primera d'aquestes dues retòriques és la que s'explica a un article de BMJ. Són dades conegudes mostrades d'una altra manera. A la meva conferència a la Reial Acadèmia de Medicina del novembre passat ja ho vaig reflectir. La segona retòrica, la del valor, també apareix d'esquitllada a l'article, però sens dubte és el tema que es vol enfatitzar en aquest moment.
El relat dels preus alts per recuperar inversió ja sap tothom que no se sosté per enlloc, i no cal donar-hi més voltes. I quan entrem en la retòrica del valor aquí hi podem trobar de tot, regles de rescat incloses. Per tant entrem a un camp minat, terres fangoses i sense cap concreció d'on anirem a parar.
Al final donen recomanacions al govern:
• Making national patent systems more stringent to avoid rewarding chemical novelty and inventiveness independent of added therapeutic value
• Clear communication by public health authorities to lay out health needs focused research and development priorities and the strategic use of public research funding to support them
• Smarter allocation of public research funds with retention of (partial) ownership that can be leveraged to pursue public health objectives, including affordable pricing
• Raising evidence standards for market authorisation by requiring companies to conduct comparative clinical trials designed to establish added therapeutic value whenever possible, and
• Reforming pricing and reimbursement systems to reward companies that develop drugs that deliver clinical benefit and discourage me-too and evergreening strategies
Llegiu-vos l'article i guardeu-lo. S'apropen els medicaments per l'hemofília i en sentirem a parlar dels seus preus i de l'impacte pressupostari. L'argument no serà la recerca, serà el valor que pressumptament aporten. Veig nirvis per totes bandes.

Banksy