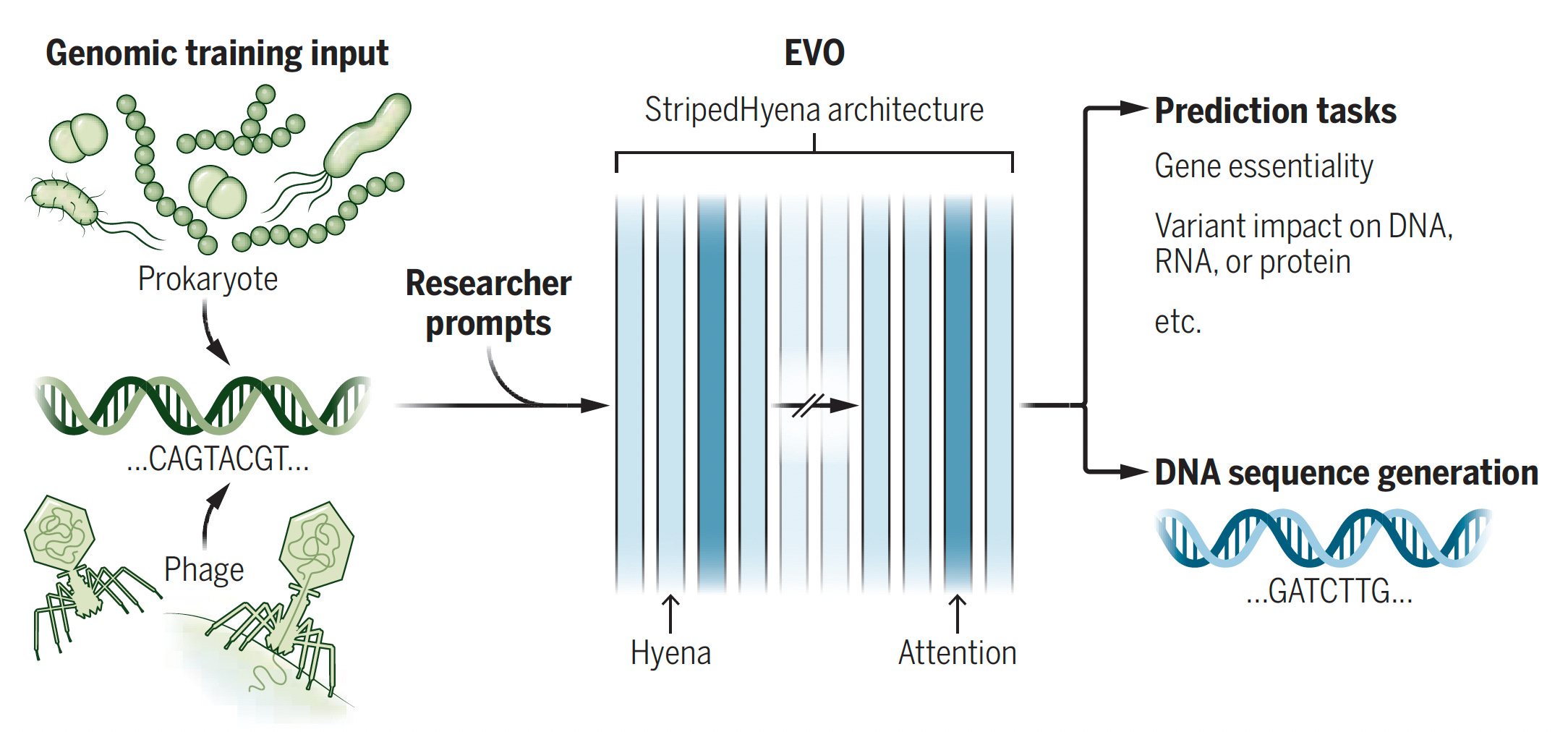
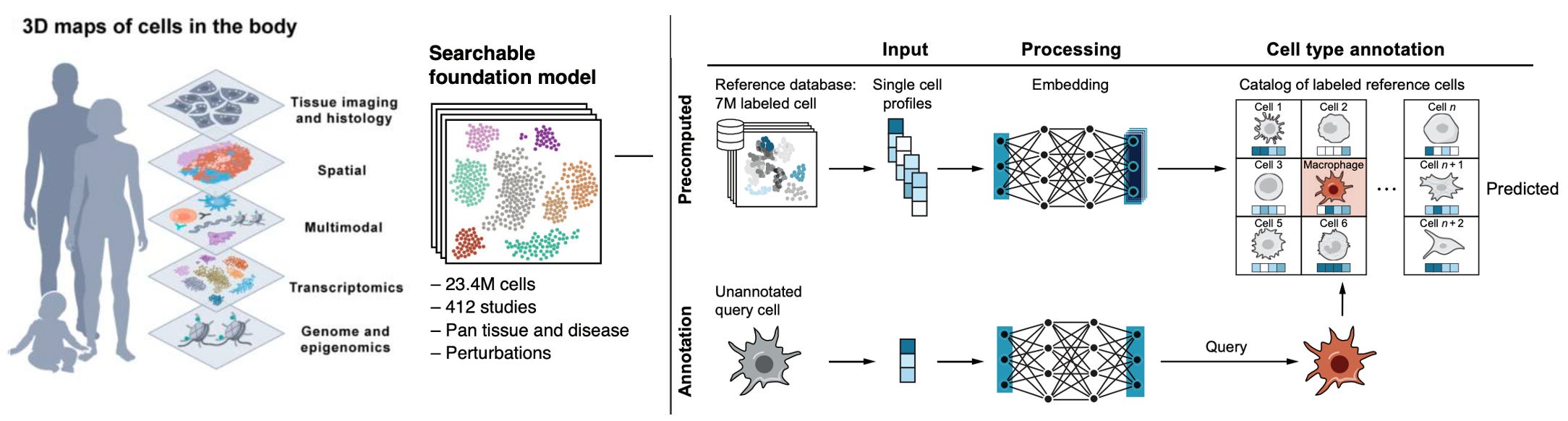
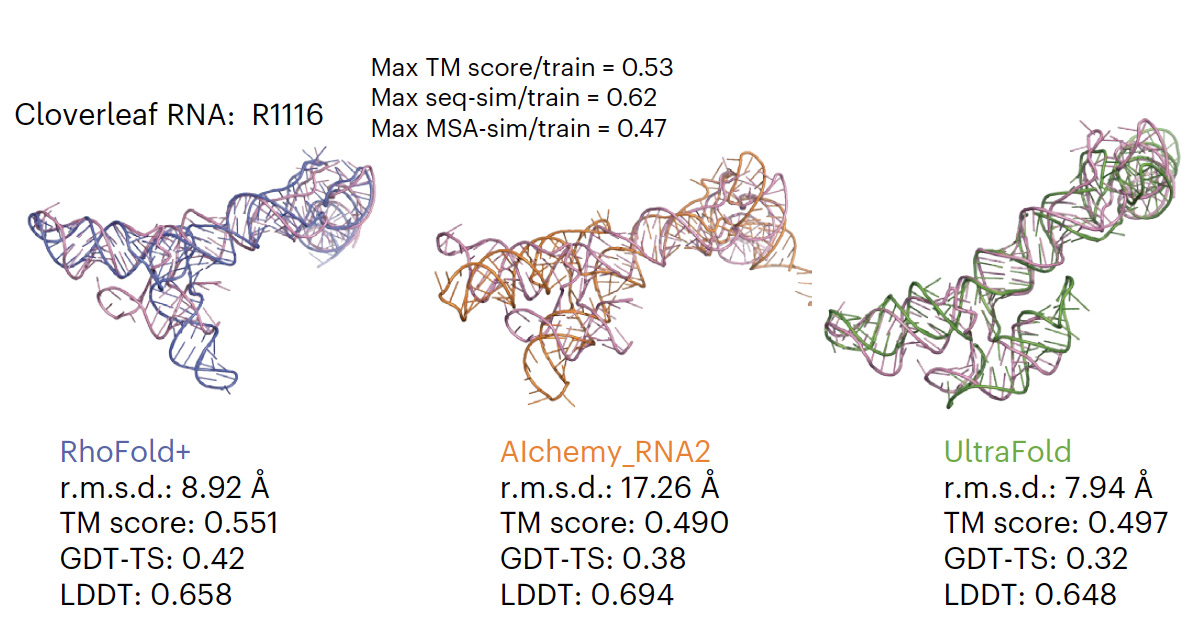
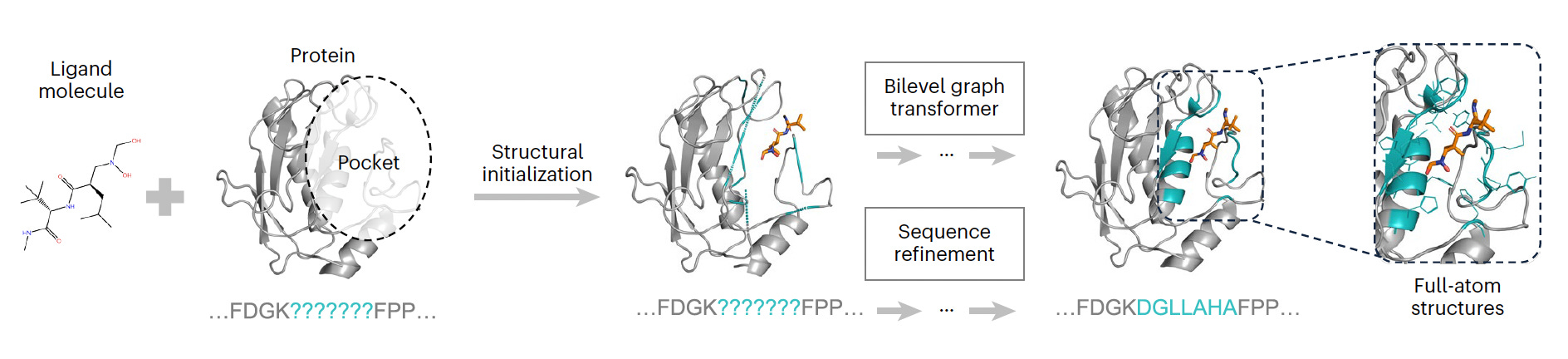
Atlas of the plasma proteome in health and disease in 53,026 adults
Hem arribat a desxifrar el 93% del Proteoma humà. Aquesta és una fita molt rellevant. I si ja havia parlat algunes vegades de l'Atles Cel·lular, ara toca fixar-nos amb el proteoma.
El més important és relacionar les proteïnes amb malalties i trets de salut. Això és el que s'ha presentat recentment a Cell i aquí resumeixo les 27 pàgines amb IA.
Aquest article presenta un estudi proteòmic a gran escala que relaciona 2.920 proteïnes plasmàtiques amb 406 malalties prevalents, 660 malalties incidentals i 986 trets relacionats amb la salut en 53.026 adults del Biobanc del Regne Unit. L'estudi pretén crear un atles proteòmic complet per a la medicina de precisió, identificant biomarcadors per al diagnòstic i la predicció de malalties, i també possibles objectius terapèutics i oportunitats de reposicionament de fàrmacs.
A continuació es detallen les troballes i els mètodes principals de l'estudi:
**Associacions proteïna-malaltia:** Es van identificar **168.100 associacions proteïna-malaltia** i **554.488 associacions proteïna-trets**. Més de 650 proteïnes es van compartir entre almenys 50 malalties, i més de 1.000 van mostrar heterogeneïtat per sexe i edat. Algunes proteïnes van mostrar un potencial prometedor en la discriminació de malalties (àrea sota la corba [AUC] > 0,80 en 183 malalties).
**Anàlisi de regressió:** Es van utilitzar models de regressió logística per a les malalties prevalents i models de regressió de riscos proporcionals de Cox per a les malalties incidentals per investigar la relació entre els nivells de proteïnes plasmàtiques i les malalties.
**Proteïnes causals:** Mitjançant la integració de dades del locus de trets quantitatius de proteïnes (pQTL), es van determinar **474 proteïnes causals**, cosa que va comportar **37 oportunitats de reposicionament de fàrmacs i 26 possibles objectius terapèutics** amb perfils de seguretat favorables.
**Pleiotropia:** La majoria de les proteïnes van mostrar associacions multifenotípiques, amb algunes com la **GDF15** i la família del **TNF** que es van associar amb moltes malalties. Per exemple, **GDF15** es va associar amb la majoria de les malalties, incloses 205 prevalents i 397 incidents.
**Anàlisi de subgrups:** Es van dur a terme anàlisis de subgrups per sexe i edat per revelar associacions específiques. Es van trobar 37.979 i 22.911 associacions específiques per sexe en les associacions proteïna-malaltia incidentals i prevalents, respectivament.
**Anàlisi de sensibilitat:** Les anàlisis de sensibilitat van revelar que la majoria de les associacions proteïna-malaltia van seguir sent significatives després d'ajustar per la comorbiditat.
**Funcions biològiques:** L'anàlisi d'enriquiment funcional va revelar que les vies relacionades amb el sistema immunitari s'enriqueixen principalment en diverses malalties. Les vies relacionades amb la unió de TNF als seus receptors fisiològics van ser les més freqüents entre les malalties.
**Classificació de malalties:** Es va aplicar una agrupació jeràrquica per agrupar les 660 malalties en 40 grups basats en les magnituds de les associacions proteïna-malaltia. Les malalties amb similituds es van agrupar i van mostrar característiques biològiques singulars.
**Diagnòstic i predicció:** La integració de les proteïnes plasmàtiques a models demogràfics va millorar significativament la precisió diagnòstica i predictiva de moltes malalties.
**Randomització mendeliana (MR):** L'anàlisi MR va identificar proteïnes causals que contribueixen a la patogènesi de les malalties i proteïnes que poden ser conseqüència de determinades malalties. Per exemple, la proteïna **GDF15** es va associar causalment amb diverses malalties autoimmunes.
**Validació i reposicionament d'objectius terapèutics:** Es va identificar que moltes proteïnes associades a la malaltia coincidien amb el genoma utilitzable per a fàrmacs, cosa que indicava possibles objectius terapèutics. Es van descobrir **37 oportunitats de reposicionament de fàrmacs** per a 25 objectius terapèutics establerts.
**Interfície web:** Es va desenvolupar una eina web interactiva per explorar els resultats.
Aquest estudi proporciona un recurs de gran escala per a la investigació futura sobre el paper de les proteïnes en la patogènesi, detecció, diagnòstic i tractament de les malalties humanes. A més, l'estudi destaca el potencial de la proteòmica plasmàtica per a la implementació de la medicina de precisió.
PS Alphafold3, la gran eina.